Défis pour la sécurité à défaut unique dans les dispositifs médicaux
TÜV SÜD examine les obstacles au développement de dispositifs médicaux électriques ou électroniques sûrs.

Les concepteurs et développeurs de dispositifs médicaux sont conscients de la pertinence et des risques liés aux défauts uniques, qui doivent être évités dans tous les états de fonctionnement. Cependant, la dynamique de développement, les progrès technologiques et le cadre normatif nécessitent une expertise approfondie, notamment pour les équipements innovants.
D’un point de vue réglementaire et technique, il est très clair pourquoi la sécurité de premier défaut doit être assurée dans les équipements médicaux électriques, électroniques et électroniques programmables (systèmes E/E/PE). Par exemple, la dose de médicament délivrée par une pompe à perfusion ne doit jamais être trop élevée ou trop faible, et une couveuse néonatale pour nouveau-nés prématurés doit maintenir en toute sécurité et fiabilité la température dans des limites étroites, sans jamais dépasser ou descendre en dessous de ces limites, même en cas de un dysfonctionnement.
Cependant, dans la pratique, les types d’équipements posant des défis supplémentaires aux fabricants sont pour la plupart beaucoup plus complexes, notamment les appareils à rayons X, les scanners IRM ou les machines d’oxygénation par membrane extracorporelle (ECMO). Pour aggraver les choses, les normes techniques ne sont pas toujours univoques. Ceci est également précisé par la fiche d’interprétation CEI 60601-1:2005/AMD1:2012/ISH1:2021, publiée en mars 2021 par la Commission électrotechnique internationale (CEI).1
Sommaire
Moins de marge de manœuvre pour l’interprétation
Le document souligne que les normes de base n’apportent pas de réponses satisfaisantes à des questions importantes telles que : Comment les fabricants peuvent-ils garantir la sécurité fonctionnelle de leurs équipements médicaux et la documenter conformément aux exigences d’accès au marché ? À quelles exigences les logiciels, les systèmes de commande et les dispositifs de sécurité doivent-ils répondre ? Quelles architectures système et de sécurité sont adaptées pour continuer à maintenir les fonctions clés de l’équipement et protéger la sécurité des patients et des utilisateurs même en cas de panne ?
Le cadre juridique applicable dans l’Union européenne est établi par le règlement sur les dispositifs médicaux (UE) 2017/745, également appelé MDR.2 Il décrit toutes les exigences qui doivent être remplies par les fabricants ou les distributeurs (par exemple, les importateurs) d’équipements médicaux avant qu’ils ne soient autorisés à mettre leurs produits sur le marché européen. Cependant, les exigences liées à la sécurité fonctionnelle restent relativement génériques. Par exemple, à l’annexe I, le règlement inclut l’exigence suivante pour la sécurité en cas de premier défaut : « En cas de condition de premier défaut, des moyens appropriés doivent être adoptés pour éliminer ou réduire autant que possible les risques ou la dégradation des performances qui en résultent. » 3
Deux options pour la conception et l’architecture des dispositifs médicaux
Fondamentalement, cela se traduit par deux options pour la conception du produit et l’architecture du système. Soit la conception du produit et l’architecture du système sont telles que la possibilité de défauts uniques est complètement éliminée, soit les fabricants effectuent des analyses de risques, garantissant ainsi que (1) l’apparition d’un défaut est hautement improbable ou (2) que ses conséquences sont mineures ou de gravité négligeable. Selon la complexité d’un appareil, l’exclusion complète des défauts uniques peut être impossible.
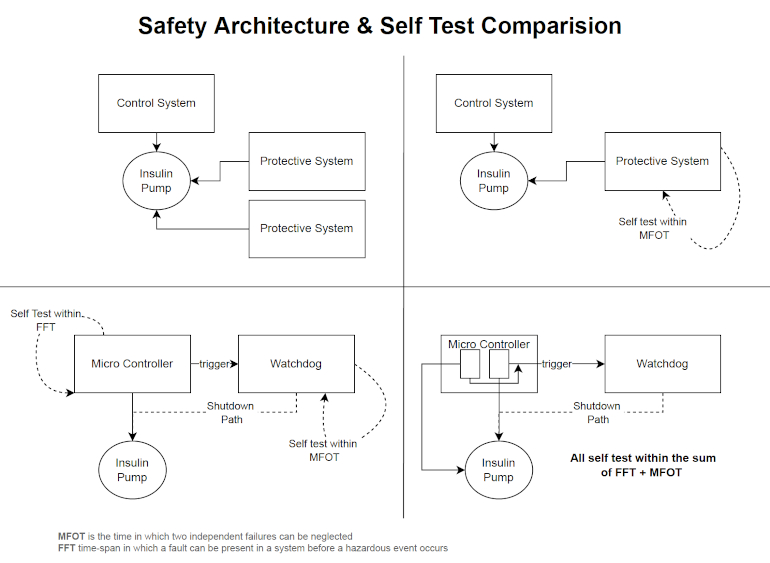
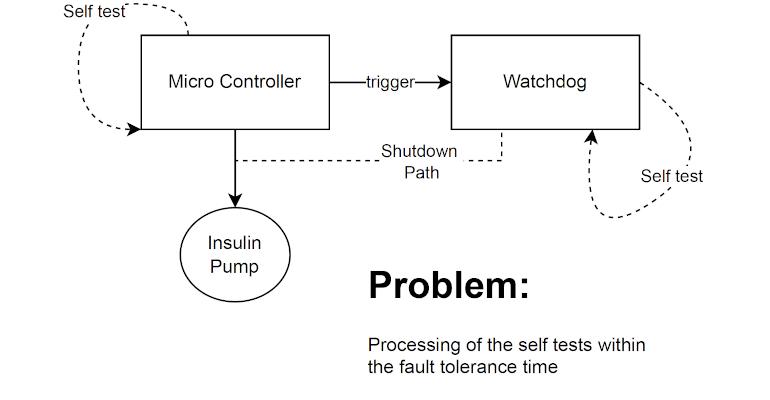
La série de normes CEI 60601 décrit ces deux options plus en détail.4 La norme présente l’état de l’art en matière d’équipements médicaux et définit les exigences de base en matière de sécurité fonctionnelle et de performances essentielles, en particulier dans la partie 1. Cependant, elle ne fournit pas non plus aux concepteurs et aux développeurs d’exigences ou d’explications explicites sur la manière dont la sécurité en cas de défaut unique des un dispositif médical peut être mis en œuvre, testé et documenté conformément aux exigences légales pour accéder aux marchés respectifs. Encore une fois, la norme ne fait référence qu’à la gestion des risques selon la norme ISO 14971 dans ce contexte.5
Elle ne décrit pas non plus de manière suffisamment détaillée les sources potentielles de dysfonctionnements, y compris les vices cachés, ni les mesures possibles pour les prévenir. Ces défauts cachés restent par définition non détectés, provoquant des dysfonctionnements discrets des dispositifs de sécurité et des systèmes de surveillance. En cas de défaut, ces dispositifs et systèmes de sécurité ne fonctionnent alors pas correctement et ne déclenchent pas, par exemple, d’alarme si un seul défaut réel se produit. Certes, la fiche d’interprétation émise par la CEI montre comment le concept de sécurité par défaut unique est appliqué aux performances essentielles et à la fonction clinique. Il comprend également les exigences relatives à la documentation (Sections bb 1 à bb 6) et à son examen. Cependant, ce document ne fournit pas non plus de réponses satisfaisantes aux questions sur la manière dont la sécurité à un seul défaut peut être obtenue et testée en tenant compte des « défauts latents ».
Trouver des solutions possibles dans d’autres secteurs
Compte tenu de ce qui précède, les fabricants, concepteurs et développeurs d’équipements médicaux sont bien avisés d’avoir une vue d’ensemble des bases et des principes de la sécurité fonctionnelle au-delà des normes spécifiques à l’industrie et à la technologie. Des conseils supplémentaires, par exemple, peuvent être trouvés dans les normes de la série EN 61508. Elles servent de normes de sécurité de base pour tous les secteurs de l’industrie qui s’appuient sur des systèmes E/E/PE dans des applications liées à la sécurité où leur sécurité fonctionnelle doit être garantie à tout moment.
Dans le cas de la conception et du développement de dispositifs médicaux innovants et complexes pouvant entraîner des risques sanitaires majeurs pour les patients ou les utilisateurs en cas de panne, il peut s’avérer intéressant de faire quelques détours dans d’autres secteurs de l’industrie, où les dysfonctionnements de l’E/E Les systèmes /PE peuvent avoir des conséquences tout aussi graves (par exemple, industrie de transformation, énergie atomique ou industrie ferroviaire). Des tiers ayant une expertise et une longue expérience dans des secteurs industriels très variés liés à la sécurité, tels que TÜV SÜD, peuvent donner une impulsion significative aux concepteurs et aux développeurs et les aider à identifier toutes les sources potentielles d’erreurs, par exemple, dans l’architecture du système, logiciel, ou même un fonctionnement incorrect, et développez des solutions possibles.
Les références
- CEI 60601-1:2005/AMD1:2012/ISH1:2021 : Feuille d’interprétation 1 – Amendement 1 – Appareils électromédicaux – Partie 1 : Exigences générales pour la sécurité de base et les performances essentielles.
- Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant Directives du Conseil 90/385/CEE et 93/42/CEE.
- Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux (annexe I, 17.1).
- CEI 60601-1 Appareils électromédicaux – Partie 1 : Exigences générales pour la sécurité de base et les performances essentielles.
- ISO 14971:2020-07 Dispositifs médicaux – Application de la gestion des risques aux dispositifs médicaux.